|
医薬品開発は個々の研究者による勘や経験によるところが大きく、膨大な時間と費用を要する。日本製薬工業協会のまとめ(2001〜2005年)では、実に開発期間9〜17年、開発費用1新薬あたり約500億円、医薬品開発の成功確率15,622分の1と見積もられている。これまでに、これらを加速化する有力な方法として、計算機を用いて化合物探索を行うインシリコスクリーニングが実施されてきたが、現状の理論や性能について頭打ちの状態にあり、業界全体としてブレイクスルーとなる新規手法が待ち望まれていた。
受賞者は、医薬品開発プロセスのボトルネックの一つである医薬品候補化合物の探索過程(数百万種類の膨大な化合物の中から、活性化合物を発見する工程)の劇的効率化を目的として、従来法であるドッキング計算法とは全く概念の異なる独自の活性化合物探索計算法「相互作用マシンラーニング法」の開発を行った。相互作用マシンラーニング法は、コンピュータによるパターン認識技術を用いて、タンパク質と化合物との結合情報(ケミカルゲノミクス情報)から抽出した結合パターンに基づいて活性化合物を効率的に発見するという受賞者が世界で初めて実用化に成功した技術であり、従来型のドッキング計算法を凌駕する性能を有している。具体的には、パターン認識に基づく機械学習アルゴリズムを用いて、化合物群とタンパク質群との多種多様な相互作用パターンの統計的な法則性をルール化することにより、活性化合物を高精度に探索予測できる計算プログラムである。
従来手法では標的タンパク質の立体構造が必要で、かつ計算コストが高く、計算結果を出すのに3ヶ月程度かかっていたのに対し、新手法ではタンパク質の立体構造なしで予測が可能であり、計算コストも非常に低く数週間で結果を出すことができる。さらに新手法
は、従来手法よりも約10倍の高い予測精度と新規化学構造の発見能力を有するなど、特筆すべき性能を有している。
本技術は、平成20年3月末に京都大学「医学領域」発の創薬系ベンチャーとして事業化され、現在、多くの創薬現場で用いられている。本技術の普及により、医薬品開発のスピードアップや新薬の創出がもたらされ、結果として、国民の福祉と健康増進に貢献するものと期待される。
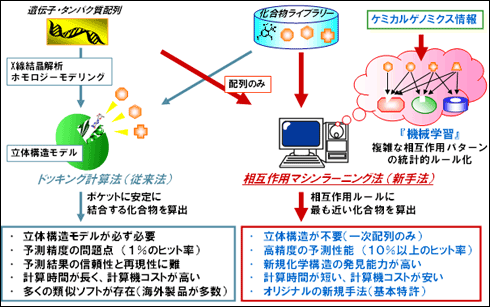
図1 相互作用マシンラーニング法(本手法:右)とドッキング計算法(従来法:左)との比較
|














